Structural Chemogenomics Group
Laboratoire d'Innovation Therapeutique, UMR7200 CNRS/Universite de Strasbourg
|
Our team aims at rationalizing and
accelerating the discovery of innovative bioactive molecules. It stands at the
interface of many disciplines (informatics, medicinal chemistry, structural
biology, pharmacology) and therefore attracts students from quite diverse areas
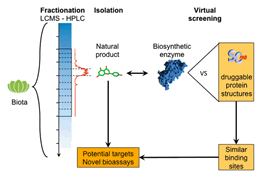
of expertise. We do perform virtual screening and medicinal chemistry
optimization in-house along three main research areas:
·
Unravelling molecular mechanisms of action
Our team has has played
a pioneering role in promoting virtual screening in Europe (Bissantz et al. 2000)
and keeps on developing
and applying in silico screening methods.
1. Databases
·
Bioinfo-DB:
repertoire of 9 million commercially available drug-like compounds from >25
suppliers (see more )
·
sc-PDB: archive
of 10 000 druggable-protein ligand complexes, used
worldwide for structure-based drug design (> 220 citations; see
more )
2. Methodological
developments
·
ChemoGen: virtual screening in cgemogenomics space with in-house devloped kernels (see
more)
·
Grim: description of protein-ligand interaction
patterns by graphs (see
more), recently
ranked 2nd/43 for predicting binding poses in the D3R Grand
Challenge 2015 (see
more)
·
Profiler: off-target prediction method, using a mix
of ligand-based (2D and 3D similarity search, pharmacophore search) and
structure-based docking approaches (see
more)
· RLPharm: automated method to define protein-ligand based pharmacophores, developed in collaboration with BIOVIA (see more). Has led to a unique collection of > 140K pharmacophores for studying either off-target activity or for drug repurposing (see more)
·
VolSite: automated
detection of protein cavities and druggability
estimate (see
more)
3. Recent in silico
screening applications
We have undertaken 14 in silico
screen campaigns over the last 5 years, mostly with the objective to identify
the very first ligands of novel targets of high pharmacological value.
a Pharmacophoric search, bpositive allosteric regulator, c chemogenomic screening, d negative allosteric
regulator, eprotein-protein interface stabilisers
Altogether, we have identified 42
hits which were fully confirmed experimentally and led to medicinal chemistry
hit to lead optimization programs (TrKB, Stat3, SmNACE, CD38, FLT3, intéine
a5b1, Mfd, Smoothened). Among
the most interesting hits, we have notably found:
·
the very first nanomolar extracellular
inhibitor of the TrkB tyrosine kinase receptor (ANA-12)
with potent in vivo antidepressant and anxiolytic properties;
·
the first TrkB positive
allosteric modulator (TB001), inducing a TrkB
specific neurite growth with potential applications in neurodegenerative
diseases;
·
the first extracellular inhibitor of the FLT3
tyrosine kinase receptor (BDT001) with potent anti-hyperalgesic
properties in rodent models of neuropathic pain;
·
inhibitors of the Smoothened oncogene, targeting the homodimeric form of the receptor, with potent anti-tumoral activities
·
inhibitors of the Mfd virulence factor, with antibacterial properties in multi
drug resistant infections. 
Pharmacodynamic and
pharmacokinetic properties of our in silico hits are optimized in-house by our
medicinal chemistry group. Here are 3 examples:
1. TrkB
extracellular inhibitors
Trkb is the
tyrosine kinase receptor for BDNF (Brain derived neurotrophic factor), a potent
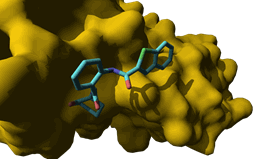
neurotrophin essential for neuronal growth. Our Trkb extracellular inhibitor (ANA-12) is potent (IC50 = 4.3
nM), selective (vs. Trka1 and TrkC)
and exhibits potent antidepressant and anxiolytic properties in rodents when administered
i.p. at the dose of 0.5 mg/kg. Within the framework
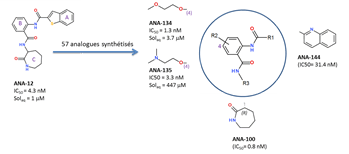
of an ANR grant (TrkBDNFarmod: ANR 11-BSV4-017-02, we have
synthesized 57 analogs by varying the heteroaromatic
ring A, the three amide bonds, the core aromatic ring B and the lactame C.
Collaborations: M. Cazorla
(Grenoble Institute of Neurosciences, Grenoble, France)
L.
Lanfumey (Psychiatry and Neurosciences Center, Paris)
M.
Hamon (Psychiatry and Neurosciences Center, Paris)
Observed structure-activity
relationships indicate that: ·
a position (position 4) of the B ring that is
tolerant to the substitution with solubilizing moieties without altering in
vitro potency; ·
2 out of the 3 amide bonds cannot be altered; ·
ring A is very susceptible to chemical variations; ·
the binding of ANA-12 to TrkB
is stereoselective: (R)-isomer being 1000
times more potent than the (S)-isomer; ·
a good permeation of the blood-brain barrier for some
analogs (e.g. ANA-135)
2. FLT3 extracellular inhibitors
FLT3 is the receptor tyrosine kinase
for the FL cytokine. Beyond its well established role in promoting cell
differentiation in hematopoietic cells, we have discovered the unique property
of FLT3 to behave as a main hub for triggering chronic pain upon nerve injury. Within
the framework of a maturation program (INSERM-Transfert, SATT AxLR-Montpellier),
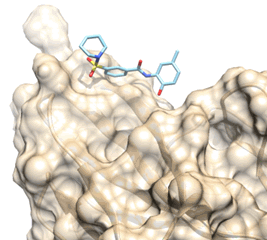
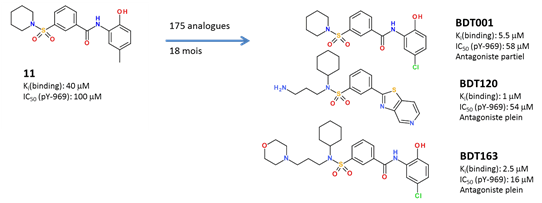
one in silico hit (compound 11) previously identified to prevent the extracellular
binding of the FL cytokine to its FLT3 tyrosine kinase receptor has been
optimized by medicinal chemistry.
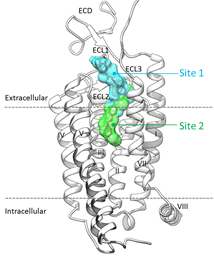
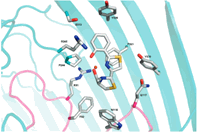
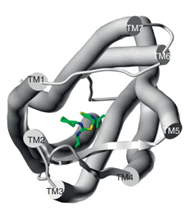
Proposed binding mode of the In silico Hit to the FLT3 receptor (tan surface)
Chemical variations on FLT3 inhibitors We notably:
·
enhanced in vitro potency (x40);
·
switch from partial to total functional antagonism
·
enhanced aqueous solubility(x400);
·
selectivity for off-targets
·
enhances metabolic stability
Optimized analogues (e.g. BDT001, BDT120, BDT169) exhibit
strong anti-hyperalgesic properties in rodent models (CCI,
SNL, SNI) of neuropathic pain, at low
doses (2.5-5 mg/kg i.p.). The chemical series is protected by a patent
(WO 2016016370 A1) Collaborations: J. Valmier
(Institute of Neurosciences, Montpellier, France)
3. CD38 inhibitors
CD38 is a cell surface receptor acting as activation
biomarker of immune cells. It is implicated in numerous cell signaling mechanisms
among which survival of heart cells. Within the framework of a
collaboration with SERVIER Laboratories, we have
synthesized 59 analogs of our initial in silico hit (ICD4) and discovered
potent human CD38 inhibitors (IC50 = 900 nM). The most
promising compounds are currently evaluated for their effects on cardiomyoctes. We separately optimized another CD38
inhibitor selective for the Schistosoma mansoni receptor (see
more )
Collaborations: A. Wagner, H. Steffner-Muller (UMR7199, Illkirch,
France)
Collaborations: A.E. Alewijnse
(Academic Medical Center, Amsterdam,The
Netherlands.) D. Tourwé (Vrije
Universiteit, Brussels, Belgium)
J.L.
Galzi, I. Schalk (UMR 7242
CNRS, Ecole Supérieure de Biotechnologie de Strasbourg, Université de
Strasbourg)
T.
Durroux, B. Mouillac, G.
Guillon, J.P. Pin (Institut de Gémique Fonctionnelle ,
Montpellier, France)
M. Ruat (Alfred Fessard Institute of Neurobiology,Gif Sur Yvette, France) B. Lagane, F. Arenzana-Seisdedos (Pasteur Institute, Paris)
R. Quinn (Eskitis Institute
for Drug Discovery, Griffith University, Brisbane, Australia.)
|